Elektryczny moment dipolowy jest wektorową wielkością fizyczną, która opisuje dipol. Dipol jest układem ładunków elektrycznych o tej samej wartości lecz posiadających przeciwny znak. Elektryczny moment dipolowy μ wyraża się za pomocą równania:

Jednostką układu SI, za pomocą której wyrażany jest elektryczny moment dipolowy jest C·m (kulomb·metr), gdzie C = 6.24150·1018 e. Często stosowaną zamiennie jednostką elektrycznego momentu dipolowego jest debaj (D), który ma wartość równą 3.33564·10-30 C·m.
Energia potencjalna oddziaływania dwóch dipoli elektrycznych o momentach dipolowych μ1 i μ2, które znajdują się w odległości r wyraża się równaniem (εo – przenikalność dielektryczna późni):

dla oddziaływania krańców dipoli o różnoimiennym ładunku, oraz:

Oddziaływanie jon-dipol występuje pomiędzy jonem a cząsteczką polarną. Energię potencjalna oddziaływań pomiędzy jonem o ładunku q będącym w odległości r od dipola μ opisuje równanie:

Jeżeli cząstka posiadająca ładunek, na przykład kation zbliży się do cząstki obojętnej, to może zmienić jej gęstość elektronową, cząstka ta uzyskuje wtedy indukowany moment dipolowy. Energię potencjalną oddziaływania obydwu cząstek opisuje równanie:


Również cząsteczka posiadająca moment dipolowy może powodować w cząsteczce obojętnej indukowany moment dipolowy. Energia potencjalna takiego oddziaływania opisana jest równaniem:

gdzie α jest polaryzowalnością cząsteczki niepolarnej, μ jest momentem dipolowym cząsteczki polarnej.
Energia potencjalna dla oddziaływań jon-dipol indukowany i dipol-dipol indukowany nie zależy od temperatury. Oddziaływania jon-dipol indukowany i dipol-dipol indukowany są słabe w porównaniu z oddziaływaniami jon-dipol.
Polaryzowalność jest to wielkość fizyczna określająca zdolność odkształcenia rozkładu ładunku w cząsteczce pod wpływem zewnętrznego pola elektrycznego, definiuje się ją jako współczynnik proporcjonalności pomiędzy natężeniem tego pola a elektrycznym momentem dipolowym przez to pole indukowanym. Zależność tą opisuje równanie:
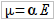
gdzie E jest natężeniem zewnętrznego pola elektrycznego, α jest polaryzowalnością. Polaryzowalność jest na ogół wielkością anizotropową tj. zależną od orientacji osi cząsteczki względem kierunku pola.
Zmiana polaryzacji powoduje powstawanie indukowanego indukowanego elektrycznego momentu dipolowego. Całkowita polaryzowalność α jest sumą polaryzowalności elektronowej αe, jonowej αa i polaryzacji orientacyjnej (dipolowej) αo:


Polaryzowalność jonowa αj jest związana z przesunięciem w różne strony jonów dodatnich i ujemnych w polu elektrycznym, wskutek czego w sieci krystalicznej powstaje elektryczny moment dipolowy. Polaryzowalność związków jonowych opisuje równanie:

Polaryzowalność orientacyjna (dipolowa) αo związana jest ze zjawiskiem ustawienia przez zewnętrzne pole elektryczne trwałych dipoli w substancji polarnej zgodnie z kierunkiem linii sił pola. Polaryzowalność orientacyjną dla cząsteczek posiadających trwały moment dipolowy określa równanie:

Polaryzowalność orientacyjna występuje dla niskich częstości, do mikrofalowych, natomiast polaryzowalność elektronowa występuje dla najszerszej granicy częstości od zakresu widzialnego do nadfioletu. Wysokie częstości drgań powodują, że dipole nie nadążają za zmianami pola elektrycznego. Elektrony są najlżejsze i nadążają za tymi zmianami, stąd dla częstości optycznych występuje wyłącznie polaryzacja elektronowa. Do opisu ilościowego polaryzacji dielektryka stosuje się wektor polaryzacji określony równaniem:

Z polaryzowalnością ściśle wiążą się wielkości takie jak przenikalność dielektryczna oraz współczynnik załamania światła.
Dla atomów, których elektrony są słabiej związane z jądrem polaryzowalność jest większa, nierzadko elektrony walencyjne mają największy wkład do polaryzowalności, tak więc im większy atom, tym większe prawdopodobieństwo jego polaryzacji. Dla cząsteczki indukowany moment dipolowy jest wielkością tensorową ponieważ nie jest on równoległy do natężenia pola elektrycznego. Dzięki istnieniu zjawiska polaryzowalności możliwe jest wykonanie widm spektroskopowych cząsteczek, które nie posiadają trwałego elektrycznego momentu dipolowego – zajmuje się tym spektroskopia Ramana.
Elektryczny moment dipolowy obliczany jest metodami chemii kwantowej jako średnia wartość momentu dipolowego w danym stanie, zgodnie z I postulatem mechaniki kwantowej, który mówi o tym, że stan cząstki określa funkcja falowa:

zależna od położenia x i czasu t. Operator momentu dipolowego i sam moment dipolowy nie zależą od wyboru układu współrzędnych tylko w przypadku molekuły obojętnej.
W metodzie Hartree-Focka elektryczny moment dipolowy jest sumą momentu dipolowego jąder i elektronów. Moment dipolowy jądra oblicza się stosując przybliżenie Borna-Oppenheimera, a moment dipolowy elektronów zgodnie z regułami Slatera-Condona. Polaryzowalność dipolową można obliczyć dwiema metodami:
- metodą sumy po stanach – polaryzowalność dipolową oblicza się stosując rachunek zaburzeń. Równanie energii n-stanu cząstki w jednorodnym polu elektrycznym zawiera człony energii cząstki, na którą nie działa pole elektryczne, człon energii oddziaływania pola elektrycznego z trwałym momentem dipolowym cząsteczki oraz energię oddziaływania pola elektrycznego z indukowanym momentem dipolowym
- metodą pola skończonego – wykonuje się obliczenia energii a z niej polaryzowalności, następnie wybiera się układ współrzędnych i przykłada pole. Realizacją sposobu tych obliczeń jest dodanie do hamiltonianu członu -μF, czyli uwzględnienie położenia układu w jednorodnym polu elektrycznym. Pewnym paradoksem obliczeń teoretycznych polaryzowalności jest fakt, że użycie bardzo dobrej bazy funkcyjnej nie daje wcale lepszych wartości, wręcz przeciwnie. Powodem jest dodany do hamiltonianu operator oddziaływania z polem elektrycznym. Tak więc obliczenia polaryzowalności w niezbyt dobrej bazie funkcyjnej dają przeważnie dość dobre wartości. Tak jest w przypadku obliczeń wariacyjnych. W przypadku obliczeń perturbacyjnych do drugiego rzędu sytuacja ta nie pojawia się. Poprawka trzeciego rzędu może dawać już pogorszenie wyników obliczenia polaryzowalności w bardzo dobrej bazie funkcyjnej.
Literatura:
1. K. Pigoń, Z. Ruziewicz: Chemia fizyczna T. 2 Fizykochemia molekularna, Wydawnictwo Naukowe PWN, 2011
2. L. Piela: Idee chemii kwantowej, Wydawnictwo Naukowe PWN, 2011

